Mutation und Reparatur
Mutationen sind vererbbare Veränderungen
der genetischen Information. Diese Information ist in allen lebenden Zellen in
Form der Deoxyribonucleinsäure (DNA), bzw. bei einigen Viren auch in Form der
Ribonucleinsäure (RNA), gespeichert. Die Aufrechterhaltung der Sequenzabfolge
dieser Information und die Weitergabe an die Nachkommen ist ein zentraler Punkt
im Leben eines jeden Individuums. Mutationen stellen aber auch die notwendige
Grundlage der Evolution dar, denn sie führen zu genetischen Unterschieden
zwischen Individuen einer gegebenen Art, an denen die Selektion ansetzen kann.
Vorteilhafte Mutationen bewirken eine bessere Anpassung an die
Umweltbedingungen, verbessern dadurch die Überlebenschancen des Trägers und
werden an die Nachkommen weitergegeben. Nachteilige Mutationen hingegen können
sich nicht durchsetzen. Mutationen ohne klaren Vor- oder Nachteil werden in der
Regel toleriert und führen zu einer Variabilität der Arten (Evolutionstheorie
von Charles Darwin).
Natürliche Mutationen sind ungerichtet: sie
entstehen aus der einfachen Tatsache heraus, dass die DNA ein labiles
Makromolekül ist, oder aber durch Einwirkungen aus dem Innern der Zelle oder
aus ihrer Umwelt. Eine gerichtete Mutagenese kann heute durch den Einsatz
gentechnischer Methoden im Labor erreicht werden.
1.
Arten von Mutationen
Mutationen werden klassischer Weise in drei
Arten unterteilt:
·
die Genommutationen, die zu einer
veränderten Anzahl der Chromosomen führen,
·
die Chromosomenmutationen, die zu
Veränderungen der Chromosomenstruktur- oder –form führen, und
·
die Gen- oder Punktmutationen, auch intragenische Mutationen genannt, bei
denen ein Nucleotid oder einige wenige aufeinander folgende Nucleotide
verändert sind.
Als Genommutationen
werden Mutationen bezeichnet, bei denen der gesamte Chromosomensatz verändert
vorliegt. Das bekannteste Beispiel ist die Trisomie 21, auch Mongoloismus
genannt. Sie fällt unter die Aneuploidien,
genauer unter die Hyperploidien,
d.h. die Vervielfältigungen einzelner Chromosomen. Das Gegenteil dazu ist die Hypoploidie, die verminderte Stückzahl
einzelner Chromosomen. Bei einer Vervielfältigung des gesamten
Chromosomensatzes spricht man von Polyploidie
und dann dementsprechend von triploiden (3n), tetraploiden (4n) usw. Zellen.
Liegt nur ein Chromosomensatz vor, so ist dies eine Haploidie.
Zu Chromosomenmutationen
kannst du im Dokument mit der Nummer 1487 mehr lesen. Zu Genom- und
Chromosomenmutationen gibt es auch einen empfehlenswerten Link:
http://www.merian.fr.bw.schule.de/Beck/Skripten/13/bs13-22.htm
Punktmutationen basieren auf dem Austauch von Basen, der Substitution. Wird eine Pyrimidinbase
(Cytosin, Thymin) gegen eine Pyrimidinbase bzw. eine Purinbase (Adenin, Guanin)
gegen Purinbase ausgetauscht, so spricht man von Transition, wird eine Pyrimidinbase gegen eine Purinbase
ausgetauscht oder andersherum, von Transversion.
Wird der fehlerhafte Bereich nicht repariert sondern abgeschrieben, manifestiert
sich die Mutation. Auch dann kann dies ohne Folgen bleiben, da der genetische
Code degeneriert ist, d.h. dass mehrere Basentripletts für eine Aminosäure (AS)
codieren können, also bereits die ersten beiden Nucleotide eines Codons die AS
eindeutig definieren. Ein Austausch des dritten Nucleotids bleibt somit ohne
Folgen, wird als stumme oder neutrale Mutation bezeichnet. Andere
Arten der Substitution hingegen ändern die genetische Information, geben ihr
einen falschen Sinn, (missense), so
dass es im Gen-Produkt zu einem AS-Austausch kommt. Auch hier können die Folgen
unterschiedlich sein: wird z.B. eine AS mit negativ geladener Seitenkette gegen
eine andere mit ebenfalls negativ geladener Seitenkette ausgetauscht, kann die
Funktion des Proteins uneingeschränkt bleiben (konservative Mutation). Wenn
jedoch eine AS im aktiven Zentrum eines Proteins ausgetauscht wird, kann dies
zum vollständigen Funktionsverlust führen. Zwischen diesen beiden Extremen gibt
es verschiedene unterschiedlich starke Auswirkungen einer Substitution auf die
Funktionstüchtigkeit eines Proteins.
Ebenfalls zum vollständigen
Funktionsverlust führen so genannte Unsinn-
oder Nonsense-Mutationen, die dann
entstehen, wenn durch eine Substitution ein Sinn-Codon in ein Stop-Codon
umgewandelt wird, da es hierdurch zur Synthese unvollständiger
Protein-Fragmente kommt. Toleriert werden können diese Verluste nur, wenn sich
das Stop-Codon am Ende der mRNA befindet und so nur zum Verlust einiger weniger
AS führt (Darstellung der unterschiedlichen Substitutionen und ihrer
Auswirkungen in Abb.1)
Abb. 1: Substitutionen und ihre
Auswirkungen
Wildtyp ATG AAG TTT GGC TAA DNA
TAC TTC AAA CCG ATT
AUG AAG UUU GGC UAA RNA
Met –
Lys – Phe – Gly – Stop Protein
stumme Mut. ATG AAG TTT GGT
TAA DNA
TAC TTC AAA CCA
ATT
AUG AAG UUU GGU
UAA RNA
Met –
Lys – Phe – Gly – Stop Protein
Missense- ATG AAG TTT AGT
TAA DNA
Mut. TAC
TTC AAA TCA ATT
AUG AAG UUU AGU UAA RNA
Met –
Lys – Phe – Ser
– Stop Protein
Nonsense- ATG TAG TTT
AGT TAA DNA
Mut. TAC ATC AAA TCA
ATT
AUG UAG UUU AGU UAA RNA
Met – Stop Protein
In der Regel tiefer greifende Folgen als
die Substitution von Nucleotiden haben der Verlust (Deletion) und die Addition
(Insertion) von ein oder zwei Nucleotiden, die so das gesamte Leseraster
verschieben (frame shift) und das
Protein gänzlich verändern (dementsprechend hat die Deletion oder Insertion von
drei Nucleotiden „nur“ die Addition oder den Verlust einer AS zur Folge).
Abb. 2: Leseraster-Mutationen (LRM) (Wildtyp s. Abb. 1)
![]() T
T
frame shift ATG AAG TTG GCT AA… DNA
TAC TTC AAC CGA TT…
AUG AAG UUG GCU AA… RNA
Met – Lys – Leu – Ala - … Protein
2.
Häufigkeit spontaner Mutationen
Dass Mutationen zufällig und ungerichtet
auftreten, d.h. die Zelle keinen Einfluss auf die Wirkung spontaner Mutationen
hat, bewiesen 1943 Luria und Delbrück (http://www.webmic.de/fluktuationstest.htm).
Die Häufigkeit der Mutationen resultiert aus der Häufigkeit der
DNA-Schädigungen und der Effizienz der zelleigenen Reparatursysteme (s.u.).
Dabei ist die Mutationsrate bezogen auf das gesamte Genom bei unterschiedlichen
Organismen, unabhängig ob Pro- oder Eukaryot, nahezu gleich. Jedes Gen kann
durch ca. 1 Mutation/109 Bakterien pro Generation getroffen werden.
Dabei beruhen im Allgemeinen etwa 70 % der Mutationen auf Substitutionen, die
übrigen Mutationen auf Deletionen oder Additionen von ein oder mehreren
Nucleotiden, also auf frame shifts. Ein kleiner Teil wird auch durch Transpositionen
verursacht, auf die später noch eingegangen werden soll.
Mutationen treten aber nicht an allen
Stellen innerhalb eines Genoms gleich häufig auf. Es gibt bestimmte Bereiche,
so genannte hot spots, die häufiger
durch Mutationen betroffen sind als andere. Dies sind Abschnitte, die reich an
desaminierbarem 5-Methylcytosin (näheres wird später noch beschrieben) sind und
Sequenzwiederholungen, bzw. kurze palindrome Sequenzen, d.h. Sequenzen, die
gegenläufig gleich sind. Warum dies so ist, wird später noch deutlich werden.
3. Ursachen für Mutationen
a) spontane
Mutationen
·
Falscheinbauten
Die DNA-Synthese im Zuge der Replikation (Replikation)
ist sehr genau, da die DNA-Polymerase (DNA-Polymerase) sehr genau arbeitet und
zusätzlich eine 3’-5’-Exonuclease-Aktivität besitzt, die eventuelle
Falscheinbauten sofort wieder entfernen kann. Dennoch können Fehler auftreten. Watson und Crick gingen davon aus, dass die Basen in verschiedenen
tautomeren Strukturen vorliegen können (Keto-Enol-Tautomerie), in denen sie
unterschiedliche Paarungseigenschaften aufweisen könnten, so dass z.B. Thymin
in der unüblichen Enolform eher mit Guanin paaren würde. Mittlerweile wurde
aber gezeigt, dass diese tautomeren Formen unter den Bedingungen der
DNA-Polymerase-Reaktion nicht vorkommen. Vielmehr sind Fehlpaarungen auf
ungewöhnliche Konformationen des DNA-Rückgrates oder der Basen zu den
Pentoseringen zurückzuführen, was zur Ausbildung so genannter Wobble-Paare führt. Mit wobble ist
gemeint, dass eine Base in ihrer Partnerwahl schwankt, wie es z.B. aus der
Codon-Anticodon-Erkennung bei der Translation bekannt ist. Am häufigsten sind
G-T- und C-A-Wobble-Paare, gefolgt von G-A-Wobble-Paaren. Sehr selten gibt es
Wobble-Paare aus zwei Pyrimidin-Basen. Die Geometrie der Wobble-Paare weicht
deutlich von der der normalen Basenpaare ab, so dass in den meisten Fällen das
unpassende Nucleotid von der DNA-Polymerase abgewiesen oder spätestens von der
3’-5’-Exonuclease entfernt wird. Außerdem verzögert ein unpassendes Nucleotid
die Kettenverlängerung und schafft so Zeit für die 3’-5’-Exonuclease zum
Entfernen des Nucleotids. Verbleibt das falsch eingebaute Nucleotid dennoch in
der DNA, kommt es zu Falschpaarungen, den so genannten Mismatches. Diese können Mutationen nach sich ziehen, außer sie
werden durch die postreplikative Reparatur (eine spätere Reparatur, s.u.)
erkannt und wieder korrigiert.
·
„Zerfallsreaktionen“
der DNA und die Entstehung von AP-Stellen
Da die DNA wie alle anderen chemischen
Substanzen nicht unbegrenzt haltbar ist, treten Zerfallsreaktionen wie hydrolytische
Spaltungen und Deaminierungen auf.
Bei der hydrolytischen Depurinierung werden die Bindungen zwischen den
Purinbasen und den Deoxyribose-Phosphat-Resten gespalten, wobei das
Deoxyribose-Phosphat-Rückgrat erhalten bleibt. So entstehen Apurinstellen. Apyrimidin-Stellen entstehen vor allem über den Umweg der Deaminierung von Cytosin-Resten, bei
der Uracil entsteht (Abb.3).
Abb. 3: Hydrolytische Deaminierung am Bsp.
von Cytosin
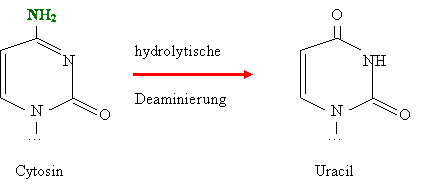
Wesentlich seltener kommt es zur
Deaminierung von Adenin zu Hypoxanthin. Zur Mutation kommt es, wenn bei der Replikation
Uracil mit Adenin paart und Hypoxanthin mit Cytosin. Um dies zu verhindern gibt
es das wirkungsvolle Reparaturenzym Uracil-DNA-Glykosylase,
das solche falschen Nucleotide erkennt, aus dem Verband der Doppelhelix
ausschwenkt und die glykosidische Bindung löst. Die Folge ist eine
Apyrimidin-Stelle. Apurin- und Apyrimidin-Stellen nennt man kurz AP-Stellen.
Hier können leicht Mutationen entstehen, da
in der Matrize ein Baustein für die Replikation fehlt, so dass komplementär im
Prinzip jedes beliebige Nucleotid eingebaut werden kann. Oft überspringt die
replikative DNA-Polymerase auch die Lücke und verursacht so eine Deletion von
einem Nucleotid. Kommt es zum Einbau einer Base wird häufiger als erwartet ein Adenin-Nucleotid
eingebaut und damit ein GC-Basenpaar in ein AT-Basenpaar überführt.
Deaminierungen können auch an anderen Basen
auftreten. Die fatalste Konsequenz hat die Deaminierung von 5-Methylcytosin zu
Thymin, da Thymin als normaler Baustein der DNA nicht vom Reparatursystem
erkannt wird. Bei der nächsten Replikation wird die Mutation fixiert, da nun
statt des GC-Basenpaares ein AT-Basenpaar erscheint. Viele Mutationsforscher
sind sich einig, dass die hydrolytische Deaminierung von 5-Methylcytosin auch einer
der wichtigsten Mechanismen für die spontane Entstehung von Genmutationen beim
Menschen ist. Dies gilt z.B. für die Bluterkrankheit, die durch eine
Fehlbildung des Gerinnungsfaktors IX verursacht wird. Vor allem CG-Folgen
weisen einen hohen Methylierungsgrad auf, so dass sie hot spots für diese Art
von Mutation darstellen. Deaminierungen können auch durch mutagene Verbindungen
wie Salpetersäure ausgelöst werden.
·
Oxidative
Schäden
Oxidative Schäden werden von
Hydroxyl-Radikalen (∙OH) verursacht, die in der Zelle über Umwege aus
Wasserstoffperoxid (H2O2), einem Nebenprodukt der
Atmungskette (siehe auch Dokument Schülerservice > Biologie >
Atmungskette ), entstehen. Der größte
Teil wird zwar durch Katalasen und Peroxidasen unschädlich gemacht, aber ein kleiner
Teil wird eben auch in reaktive
Hydroxyl-Radikale überführt. Zudem erhöhen ionisierende Strahlen – neben
der Auslösung einer Reihe anderer schwerer Schäden (s.u.) - über eine Radiolyse
aus Wasser die Bildung von Hydroxyl-Radikalen. Hydroxyl-Radikale verändern die
Struktur von DNA-Nucleotiden, am häufigsten ist dabei die Bildung von
8-Oxoguanin (8-OxoG). Mutationsforscher gehen davon aus, dass auch ohne äußere
Einwirkungen bis zu 1000 8-OxoG-Reste pro Tag im menschlichen Genom entstehen.
Der Einbau von 8-OxoG in die DNA führt zu Problemen, da ihm gegenüber bei der Replikation
entweder das normale Cytosin oder aber bevorzugt Adenin eingebaut, was zu einer
GC- nach TA-Transversion führt. Verändert sich das Guanin im freien dGTP zu
8-OxodGTP, so kann es bei der Replikation gegenüber Adenin eingebaut werden.
Die falsch eingebauten 8-OxoG-A-Basenpaare können von der 3’-5’-Exonuclease
nicht erkannt werden, weil ihre Geometrie wahrscheinlich der der normalen Basenpaare
nahe kommt. Es kommt zu einer AT-nach-CG-Transversion.
·
Ursachen
spontaner Leseraster-Mutationen (LRM)
Wie bereits erwähnt sind
Sequenzwiederholungen und kurze palindrome Sequenzen hot spots spontaner
Mutationen. Dies gilt insbesondere für Leserastermutationen (frame shifts), die
sequenzabhängig dort auftreten, wo mehrere gleiche Basenpaare hintereinander
vorkommen. Zudem begünstigen Lücken, wie sie in der Nähe der Replikationsgabel,
bei der Rekombination und bei Reparaturprozessen vorkommen, die Entstehung von Leserastermutationen.
Es kommt zur Ausbildung von Nucleotid-Schleifen, so genannten „extrahelikalen“
Nucleotiden. Genaueres erläutert die Abbildung 4 bildlich.
Abb. 4: Entstehung spontaner LRMs
ATGAACGCGCGCGCGTACG DNA mit GC-Folgen
TACTTGCGCGCGCGCATGC
ATGAACGCGCGCGCGTACG Lücke im DNA-Strang
TACTTGC GCGCATGC
![]()
![]()
![]()
![]()
![]()
GC
CG
ATGAACGCGCGTACG ATGAACGCGCGCGCGTACG
TACT TGCGCGCATGC TACTTGC GCATGC
![]()
![]() CG
CG
![]()
bei der Repl. d. unteren
![]() Stranges Deletion v. 4 BP beim Ausfüllen d. Lücke durch
Stranges Deletion v. 4 BP beim Ausfüllen d. Lücke durch
![]() Reparatursynthese Addidtion v. 2 BP
Reparatursynthese Addidtion v. 2 BP
TACTTGCGCGCATGC TACT TGCGCGCGCGCGCATGC
ATGAACGCGCGTACG ATGAACGCGCGCGCGCGTACG
·
Transponierbare
Elemente bei Bakterien
In der DNA von Bakterien gibt es
Sequenzelemente, die ihre Lage und Kopienzahl innerhalb des Genoms verändern
können. Sie stellen damit eine Art der Rekombination dar, gehen aber auch oft
mit Veränderungen der DNA-Struktur einher, v.a. mit Insertionen und Deletionen.
Den Vorgang nennt man Transposition, die Elemente selbst transponierbare
Elemente, wobei man zwischen den Insertions-Elementen
(IS-Elementen), die nur für ein oder zwei Enzyme codieren, und den komplexeren Transposons, die auch andere Gene, z.B.
für Resistenzen, umfassen können, unterscheidet. Die Insertion eines solchen
transponierbaren Elements innerhalb eines Gens kann zu dessen Funktionsverlust
führen oder aber die Genexpression verändern.
b) Induktion
von Mutationen durch Chemikalien
·
DNA-Alkylierung
Alkylierende Chemikalien sind wichtige Mutagene,
die in Experimenten eingesetzt werden, aber auch in der natürlichen Umwelt,
z.B. als Nitrosamine im Magen-Darm-Trakt von Säugetieren, vorkommen. Sie, bzw.
ihre eigentlich reaktiven Stoffwechselprodukte verändern DNA-Nucleotide an
allen Positionen, die einer chemischen Ethylierung oder Methylierung zugänglich
sind. Eine Methylierung von DNA-Nucleotiden kann auch spontan erfolgen, wobei
die Methylgruppe in der Regel von dem normalerweise bei enzymatischen
Reaktionen als Cofaktor fungierenden Methylgruppen-Überträger S-Adenosylmethionin (SAM) stammt. Die
Hauptmethylierungsprodukte dabei sind 3-Methyladenin und 7-Methylguanin. Direkte Mutationen können durch O6-Methylguanin
(O6-MeG) und O4-Methylthymin (O4-MeT)
hervorgerufen werden, da O6-MeG mit Thymin und O4-MeT mit
Guanin paart. Indirekte Mutationen
entstehen im Zuge von Reparaturprozessen, wenn nach der Entfernung der
alkylierten Nucleotide durch eine Glykosylase die entstandene AP-Stelle nicht
oder nicht schnell genug repariert wird.
·
Polycyclische
Kohlenwasserstoffe
Polycyclische Kohlenwasserstoffe (PK)
entstehen vor allem bei unvollständigen Verbrennungen wie z.B. im Tabakrauch.
Sie werden zum Teil erst durch modifizierende Enzyme in ein wirksames Agens
überführt. Die bekanntesten PKs sind Benz(a)pyren und das Schimmelpilztoxin
Aflatoxin. Eine Anheftung dieser voluminösen Moleküle führt zu einer
erheblichen Verzerrung der DNA-Struktur, was zu der Bezeichnung „unförmige
Basenmodifikationen“ (bulky adducts) führte. Diese begünstigen die Hydrolyse
der glykosidischen Bindung zwischen der modifizierten Base und der Deoxyribose
und führen so zu vermehrten AP-Stellen, die durch falsche Adenin-Nucleotide
gefüllt werden können. Außerdem blockieren die unförmigen Modifikationen die
DNA-Replikation, so dass oft lange Einzelstrang-Regionen entstehen, die den
fehlerhaften SOS-Reparaturweg auslösen (s.u.).
·
Basenanaloga
und interkalierende Substanzen
Basenanaloga sind Substanzen, die den Basen in ihrer
Struktur stark ähneln und so statt dieser in die DNA eingebaut werden können.
Sie gehen besonders oft Fehlpaarungen mit anderen Basen ein, was mit hoher
Wahrscheinlichkeit bei der nächsten Replikation zu Mutationen führt. Bekannte
Basenanaloga sind 5-Bromuracil (BU), das für Thymin eingebaut wird und
bevorzugt Wobble-Paarungen mit Guanin ausbildet, und 2-Aminopurin (2-AP), das
wie das strukturell ähnliche Adenin mit Thymin aber auch bevorzugt mit Cytosin
paaren kann.
Interkalierende
Substanzen besitzen eine
planare Struktur und sind so in der Lage, sich zwischen die Basenpaare
einzulagern. Hierdurch können sie Replikationsfehler verursachen, die in der
Regel zu LRMs führen. Bei diesen interkalierenden Substanzen handelt es sich
meist um polyzyklische Verbindungen wie Acridin-Farbstoffe oder Proflavine, die
auch im Labor zur Färbung von DNA eingesetzt werden. Andere interkalierende
Substanzen, wie das Antibiotikum bzw. Antitumormedikament Mitomycin, reagieren
kovalent mit der DNA und führen zum Teil zur Quervernetzung der DNA-Stränge,
wodurch die Replikation verhindert wird.
·
Mutationen
durch UV-Licht
Am häufigsten verursachen UV-Strahlen
Reaktionen zwischen benachbarten Pyrimidinen, bevorzugt zwischen zwei
Thymin-Resten, wodurch es zu einem Thymin-Dimer kommt, das über einen
Cyclobutan-Ring verbunden ist. Diese Reaktion kann bis zu 85% der UV-Schäden
ausmachen. Nur in etwa 10% der Fälle kommt es zu einem TC(6-4)-Photoprodukt.
Beide führen in jedem Fall zur Verzerrung der DNA-Doppelhelix.
·
DNA-Schäden
durch ionisierende Strahlen
Elektromagnetische Strahlen (Röntgen- und -Strahlen)
und korpuskuläre Strahlen (- und -Strahlen) erzielen beim
Eindringen in Zellen direkte Wirkungen, wenn sie auf ein Makromolekül treffen,
oder indirekte Wirkungen, wenn sie mit Wassermolekülen in der Zelle reagieren
und so Hydroxyl-Radikale bilden (s.o.). Die meisten Schäden dabei sind
Veränderungen in der Struktur der Basen, wie der oben beschriebene wichtigste
Schaden 8-OxoG. Andere Schäden betreffen die Struktur der DNA: Dies sind
Quervernetzungen zwischen Basen der beiden DNA-Einzelstränge sowie Einzel- und
Doppelstrangbrüche, die besonders schwerwiegende Folgen für die Struktur und
die Funktion des Genoms haben können.
4. Reparaturmechanismen
Da die Wahrung der DNA-Struktur essentielle
Bedeutung für jede Zelle hat, haben sich verschiedene Mechanismen zur
Beseitigung von Schäden entwickelt. Direkte Korrekturmöglichkeiten sind die
lichtabhängige Spaltung von Pyrimidin-Dimeren oder die Demethylierung von alkylierten
Basen. Meist aber muss der beschädigte Abschnitt durch mehrere Enzyme großzügig
aus der DNA ausgeschnitten werden. Sehr umfangreiche Schäden lösen eine
„Notreaktion“ der Zelle, die SOS-Antwort, aus.
·
Direkte
Reparatur modifizierter Basen
Bestimmte Basenmodifikationen, nämlich
UV-Schäden und bestimmte DNA-Alkylierungen, können direkt wieder rückgängig
gemacht werden. Bei der Photo-Reaktivierung
können Bakterien mit Hilfe des sichtbaren Lichts (Wellenlängen von 340-400 nm) und
des Enzyms Photolyase Pyrimidin-Dimere
in der DNA durch Spaltung des Cyclobutan-Rings wieder beseitigen. Zwei Chromophore
fungieren dabei als Cofaktoren: 5,10-Methylen-tetrahydrofolat sammelt die
Lichtenergie und überträgt Elektronen auf das Flavinadenin-Dinucleotid (FAD).
Die reduzierte Form FADH2 liefert die Elektronen für die Spaltung
des Cyclobutan-Ringes. Solche Mechanismen finden sich auch bei einigen
Eukaryoten, allerdings nicht bei Säugern.
Ein Enzym namens O6-Methylguanin-DNA-Methyltransferase (MGMT) kann in den
Zellen vieler Arten (die Untersuchungen und damit die Bezeichnungen stammen
aber wieder einmal von E.coli) sowohl O6-Guanin- als auch O4-Thymin-Alkylierungen
und die Alkylierung von Phosphaten wieder rückgängig machen. Die Methylgruppen
von O6-MeG und O4-MeT werden dabei auf die Seitenkette
eines Cysteins im carboxyterminalen Bereich der MGMT, die Methylgruppen von
Phosphaten an ein Cystein im aminoterminalen Bereich übertragen. MGMT kann die
Methylgruppe nicht wieder abgeben und wird so inaktiv. Experten hat die Frage
interessiert, was passiert, wenn die Bakterienzelle mit alkylierenden
Chemikalien überschwemmt wird, und ein Mangel an MGMT droht. Sie fanden eine adaptive Antwort des Enzyms selbst, da es
durch die übernommene Methylgruppe zu einem Transkriptionsfaktor wird, der
sowohl seine eigene Transkriptionsrate steigert als auch die der 3-Methyladenin-DNA-Glykosylase II. Diese
wird im Gegensatz zur 3-Methyladenin-DNA-Glykosylase
I nicht konstitutiv exprimiert (also ohne Regulation durch äußere Signale),
sondern erst im Zuge der genannten adaptiven Antwort. Beide Enzyme arbeiten
ähnlich wie die Uracil-DNA-Glycolsylase (s.o.), können aber nicht nur – wie die
Bezeichnung vermuten ließe – 3-Methyladenin-Basen sondern auch andere
alkylierte Nucleotide unter der Schaffung von AP-Stellen aus der DNA entfernen.
·
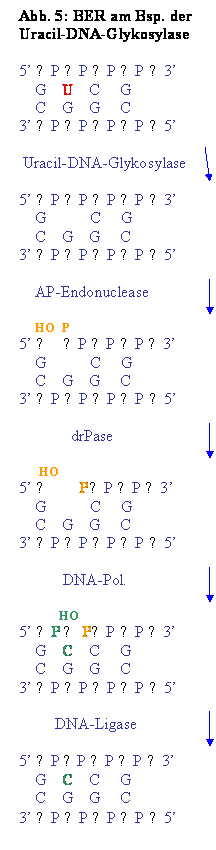 Basen-Exzisions-Reparatur (BER)
Basen-Exzisions-Reparatur (BER)
Kleinere Modifikationen an Basen, wie
Alkylierungen oder oxidative Schäden, werden durch die Basen-Exisions-Reparatur behoben. Dabei spielen Glykosylasen eine Rolle, von denen drei
bereits genannt wurden. Sie erkennen DNA-fremde Nucleotide, schwenken sie aus
dem Verband der Doppelhelix aus, lösen die glykosidische Bindung und schaffen
so eine AP-Stelle. Eine AP-Endonuclease
schneidet das Deoxyribose-Phosphat-Band 5’-wärts gleich neben der AP-Stelle, so
dass auf der einen Seite ein 5’-Deoxyribose-Phosphat-Rest ohne Base und auf der
anderen Seite eine freie 3’-OH-Gruppe entsteht. Ein Enzym namens Deoxyribophosphodiesterase (drPase)
entfernt den Deoxyribose-Phosphat-Rest. Die entstandene Lücke von einem
Nucleotid wird durch eine DNA-Polymerase, die ein Nucleotid an das 3’-OH-Ende
bindet, gefüllt, und eine DNA-Ligase verschließt das Deoxyribose-Phosphat-Band
wieder.
Der häufigste oxidative Schaden, 8-OxoG
(s.o.), wird durch die 8-OxoG-DNA-Glykosylase,
die bei E.coli auf dem so genannten MutM-Protein liegt, repariert. Dieses
MutM-Protein beinhaltet auf einer Polypeptidkette außerdem eine AP-Lyase-Funktion und eine Funktion zur
Schaffung freier 3’-OH-Enden. Die AP-Lyase trennt statt einer AP-Endonuclease
mit nicht-hydrolytischer Wirkung das Deoxyribose-Phosphat-Band an der AP-Stelle
auf, indem es die C,O-Bindung zwischen Deoxyribose und Phosphat durch eine
Eliminierungsreaktion unter Ausbildung einer C,C-Doppelbindung im
Deoxyribose-Gerüst spaltet. Die drPase-Funktion des Proteins schafft ein freies
3’-OH-Ende und die weiteren Schritte der oben beschriebenen BER folgen. Ein zu
MutM äquivalentes Protein findet sich auch bei Eukaryoten einschließlich des
Menschen.
·
Mismatch-Reparatur
Die Mismatch-Reparatur ist eine besondere
Form der BER, die sehr schnell nach dem Einbau falscher Basen erfolgt. Um ein Mismatch
reparieren zu können, muss zunächst vom System erkannt werden, welches die
falsche Base ist. Hierzu wird bei E.coli die Methylierung der GATC-Folgen (N6-Methyladenin
statt Adenin) genutzt, eine Modifikation, die eigentlich der Unterscheidung
zwischen eigener und Fremd-DNA dient. Unmittelbar nach der Replikation ist der
neu synthetisierte Strang noch nicht methyliert, was ein Signal für die
Mismatch-Reparatur ist. Im ersten Schritt bindet das MutS-Protein (Diese und
die folgenden Bezeichnungen der Proteine stammen aus der Analyse der
prototypischen Komponenten in E.coli, wobei sie sich von „mutator“ ableiten, da
Mutationen in diesen Reparatursystemen zu einer erhöhten Mutationsrate führen.)
an das unkorrekte Basenpaar, wodurch die Anlagerung von MutL und dann von MutH,
ermöglicht wird. Letzteres schneidet den neuen Strang in einer nahen unmethylierten
GATC-Folge. Die Helikase II entwindet den geschnittenen DNA-Strang, der durch
die Exonuclease I oder VII oder das RecJ-Protein abgebaut wird, abhängig davon,
auf welcher Seite des Mismatches der Strang geschnitten wurde. Die Neusynthese
zum Schließen der Lücke erfolgt über die DNA-Polymerase III, die Nucleotide an
das freie 3’-OH-Ende heftet und die Ligase, die die letzte Phosphodiester-Bindung
schließt. Das bakterielle Mismatch-Reparatursystem ist in der Lage, alle
möglichen Falschpaarungen (vielleicht mit Ausnahme von C-C-Paarungen) und
kleine Insertionen und Deletionen, bei denen ein Strang ein oder zwei
Extranucleotide enthält, zu reparieren. Außerdem kann es Mismatches in
Heteroduplex-Bereichen, die bei der Rekombination in Holliday-Strukturen
vorkommen können, entfernen.
Die eukaryotischen
Mismatch-Reparatursysteme arbeiten prinzipiell gleich, was auch zu den
Bezeichnungen MSH für MutS-Homolog usw. führte. Als bevorzugtes
Erkennungssignal dienen hier allerdings nicht die Methylierungen sondern die
freien Enden wachseneder DNA-Stränge. Ein besonderes Merkmal der eukaryotischen
Mismatch-Reparatur ist die Fähigkeit auch längere Insertionen bis zu 12 oder 16
Nucleotiden zu reparieren.
·
Nucleotid-Exzisions-Reparatur
(NER)
Die NER erkennt Störungen in der DNA- und
in der Chromatin-Struktur umso effizienter, je größer die Störungen, d.h. je
stärker die Verzerrungen (z.B. (6-4)-Photoprodukte besser als
Cyclobutan-Derivate) oder je voluminöser die Additionsprodukte (z.B.
Additionsprodukte von Benz(a)pyren besser als Methylierungen) sind.
Der Mechanismus ist bei Pro- und Eukaryoten
im Prinzip recht ähnlich, nur die beteiligten Proteine weichen voneinander ab. Der
erste Schritt ist wieder die Erkennung des DNA-Schadens, an den bei Bakterien ein Komplex aus zwei UvrA-
und einem UvrB-Protein spezifisch bindet. (Auch hier stammen die Bezeichnungen
aus Untersuchungen an E.coli, und zwar aus Untersuchungen bzgl. der
Empfindsamkeit auf UV-Strahlen; deswegen auch Uvr für uv-repair.) Nach der
Bindung wird das UvrA-Protein durch ein UvrC-Protein ersetzt und der
UvrBC-Komplex schneidet die DNA acht Nucleotide 5’-wärts und fünf Nucleotide
3’-wärts des Schadens. Eine UvrD-Helikase entfernt das geschädigte DNA-Stück. Nach
der Exzisions, dem Ausschneiden, folgen die Reparatursynthese mittels der DNA-Polymerase
I und die Versiegelung des Phosphodiester-Bandes durch eine DNA-Ligase.
Abb. 6: NER
![]()

![]()

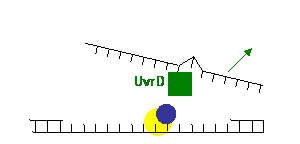
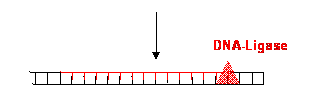
Befindet sich der Schaden im
transkribierten Strang aktiver Gene, begünstigt das Protein Mfd (mutation frequency declining),
auch bekannt als TRCF (transcription
repair coupling factor) zusätzlich deren bevorzugte Reparatur. Es verdrängt die
auf den Schaden auflaufende RNA-Polymerase und vermittelt die Bindung des
UvrA-Proteins, das die Reparatur nach dem beschriebenen Schema einleitet.
Bei Eukaryoten
wird der Schaden durch ein Protein namens XPA aufgespürt, das aber nur mit
Hilfe eines so genannten Einzelstrang-Bindeproteins an den Schaden binden kann.
XP steht für Xeroderma pigmentosum,
eine Erbkrankheit, die zu einer erhöhten Empfindlichkeit gegen Sonnenlicht und
damit zu einer hohen Wahrscheinlichkeit für das Erkranken an Hautkrebs führt. (http://www.onmeda.de/krankheiten/xeroderma_pigmentosum.html)
Bei dieser Krankheit wurden Mutationen in den Systemen der Nucleotid-Exzision
gefunden. Viele Erkenntnisse der NER bei Eukaryoten und somit auch die
Bezeichnungen stammen aus der Untersuchung solcher Patienten. Zum gebundenen
XPA-Proteine kommen dann weitere XP-Proteine hinzu, bis ein Komplex entsteht,
der den DNA-Strang 22 Basenpaare 5’-wärts und 4-6 Basenpaare 3’-wärts
schneidet. Im Komplex vorhandene Helikasen trennen das Oligonucleotid mit dem
Schaden heraus, so dass eine Lücke von 27-29 Nucleotiden entsteht, die durch
die Enzyme der DNA-Replikation (v.a. DNA-Polymerase und ) und
die Ligase wieder geschlossen wird.
·
Postreplikative
Reparatur
Schäden in der DNA, die störend auf die Replikation
wirken, können von der auflaufenden DNA-Polymerase übersprungen werden. Dadurch
entstehen lange Einzelstrangsabschnitte, die die Rekombination zwischen
homologen Molekülen anregen, woher auch die Bezeichnung rekombinative Reparatur stammt. Eine Hauptrolle spielt das Protein
RecA, das an die Einzelstrang-DNA-Enden links und rechts der Lücke bindet und
das homologe Chromosom nach homologen Sequenzen absucht. (Unter guten
Bedingungen können Bakterien rasch proliferieren und besitzen dann zwei oder
mehr Genome, da die nächste Replikationsrunde noch vor Beenden der letzten
eingeleitet wird. Dies verbessert natürlich die Chancen für eine schnelle
rekombinative Reparatur.) Nach dem Auffinden der passenden Stelle dringt der
Komplex in die Doppelhelix ein, verdrängt den homologen Strang, um an den
komplementären zu binden und ihn als Matrize für die Neusynthese zu benutzen.
Der verdrängte Strang paart sich mit dem fehlerhaften Strang. Endonucleasen
schneiden die DNA zurecht und Ligasen verschließen sie, so dass zwei komplette
Doppelhileces entstehen. Der eigentliche Schaden ist auf diese Weise nicht
behoben, sondern es wurde „nur“ die Lücke im Tochterstrang geschlossen.
·
Reparatur
von Einzel- und Doppelstrangbrüchen
Die Reparatur von Einzelstrang-Brüchen wird
durch den intakten Partnerstrang geleitet, der als Matrize dient. So wird die
Neusynthese ähnlich wie nach einer Nucleotd-Exzision durchgeführt. Die
kompliziertere Reparatur von Doppelstrang-Brüchen basiert bei Prokaryoten auf
dem Rekombinationssystem. Die Enden des gebrochenen Stranges werden auf
homologe Abschnitte eines intakten Genoms geleitet, was zur Ausbildung der
Holliday-Struktur als typischer Zwischenform bei der homologen Rekombination
führt. Auf diese Weise können wieder intakte Doppelhelices entstehen. Auch
Eukaryoten verwenden die Rekombination als Mechanismus für die Reparatur von
Doppelstrangbrüchen. Daneben gibt es bei ihnen aber auch ein zweites
Reparatursystem, die so genannte nicht-homologe
End-zu-End-Verknüpfung (non-homologous
end-joining). Die Mechanismen dieses Reparatursystems sind noch nicht ganz
geklärt. Eine Rolle spielen wohl eine Reihe von DNA-abhängigen Protein-Kinasen,
die sich als Komplex an die gebrochenen Enden binden und deren
Aneinanderlagerung begünstigen, mehrere Proteine mit Exonuclease-Aktivität, die
die DNA zurecht schneiden, sowie ein Protein namens XRCC4 (x ray repair cross
complementing) und die Ligase IV. XRCC4
plaziert vermutlich die DNA-Ligase IV so an die Enden der gebrochenen
DNA, dass eine kovalente Verbindung der DNA-Stränge und damit eine
End-zu-End-Verknüpfung möglich wird.
·
SOS-Antwort
bei Bakterien
Unter Bedingungen, die schwerste
DNA-Schäden hervorrufen, oder durch Schäden während der Replikation wird bei
E.coli eine SOS-Antwort ausgelöst. Ausgangspunkt sind lange
Einzelstrang-Regionen, wie sie beim Auflaufen der replikativen DNA-Polymerase
auf die Replikationsgabel oder als Zwischenstation bei der NER entstehen. An
diese bindet, wie oben beschrieben, das Protein RecA, das zu seinen anderen
Funktionen auch die Spaltung von drei Proteinen fördert: Lambda-cI-Repressor,
LexA-Repressor, UmuD-Protein. Für die SOS-Antwort sind die beiden letzten
entscheidend. Der LexA-Repressor blockiert über 20 Gene, die nach seiner
Spaltung im Zuge der SOS-Antwort transkribiert werden. Zu diesen SOS-Genen
gehören das sfiA-Gen, dessen Produkt die Zellteilung hemmt und so Zeit für die Reparaturen
schafft, die Gene für die Komponenten der NER, das polB-Gen für die DNA-Polymerase
II und das dinB-Gen für die DNA-Polymerase IV. Das UmuD-Protein wird als
Vorläufer vom RecA-Protein in das funktionelle Protein UmuD’ überführt, von dem
zwei Moleküle zusammen mit einem Molekül UmuC die aktive DNA-Polymerase V
bilden. Jede der drei SOS-induzierten DNA-Polymerasen kann sich (an für sie
spezifischen Schäden) anstelle der replikativen DNA-Polymerase III setzen und
im Gegensatz zu dieser die Replikation über die Schadstelle hin fortsetzen, bis
diese nach einer gewissen Strecke die Arbeit wieder aufnehmen kann. Diese DNA-Polymerasen
haben eine hohe Fehlerrate, aber sie sichern die Integrität der DNA und damit
das Überleben der Zelle.
·
Checkpoint-Kontrolle
bei Eukaryoten
Ionisierende Strahlen, UV-Strahlen oder
chemische Mutagene lösen in eukaryotischen Zellen unterschiedliche
Reaktionsketten aus. Einige davon beginnen an der Zelloberfläche und werden
über Zwischenstufen in den Zellkern übertragen, wo die Expression dutzener Gene
eingeleitet wird. Dazu gehören Proteine des DNA-Metabolismus wie DNA-Polymerase
oder Ligasen, Regulatoren des Zellzyklus, Stress-Proteine,
gewebsmodulierende Enzyme wie Collagenasen und Transkriptionsfaktoren, die
wieder andere Gene aktivieren können. Andere Reaktionen gehen von der
geschädigten DNA aus. Ein Name, der in diesem Zusammenhang oft fällt, ist der
des Enzyms Poly-(ADP-Ribose-)Polymerase,
kurz PARP, dessen Vorgehensweise zwar gut untersucht ist, nämlich die Anheftung
von verzweigten Ketten von Poly-ADP-Ribose, dessen Sinn innerhalb der
Schadensreparatur aber noch nicht bekannt ist. Eine weitere Reaktion betrifft
die eben schon genannte DNA-abhängige Protein-Kinase. Sie phosphoryliert das Protein p53, das eine wichtige Rolle in
der Regulation des Zellzyklus hat. Dieses Protein ist auch das Ziel anderer
wichtiger Kinasen, so genannter Checkpoint-Kinasen,
wie die ATM- und ATR-Kinase, die am Beginn eines komplizierten Netzwerkes
stehen, das als schadensinduzierte
Checkpoint-Kontrolle bezeichnet wird. Dieses schaltet den
Nucleotid-Exzisions-Apparat ein, aktiviert Protein-Komplexe, die zur Reparatur
von DNA-Brüchen führen und aktiviert eben auch das Protein p53. Für dessen
Aktivierung müssen mindestens drei der Protein-Kinasen zusammenkommen. Dann
fungiert p53 als Transkriptionsfaktor für Proteine, darunter p21, die
Zellzyklus und DNA-Replikation zum Halten bringen, um eine Ruhephase für
Reparaturen zu schaffen. Ist keine Reparatur mehr möglich, kann p53 auch einen
Mechanismus auslösen, der zum Absterben der Zelle führt. Dieser für die
einzelne Zelle tödliche, für den gesamten Organismus aber lebensnotwendige
Mechanismus wird als Apoptose bezeichnet. Wie wichtig dieser Mechanismus ist,
zeigt sich, wenn das Gen für p53 durch eine Mutation verändert ist. Menschen,
die mit dem mutierten Gen (offiziell als TP53 bezeichnet) geboren werden,
erkranken sehr früh an Krebs
(Li-Fraumeni-Syndrom:
http://www.mgz-muenchen.com/home.php?nid=diagnostik&uid=2&tid=7&did=77
)
Außerdem ist bei mehr als der Hälfte aller
menschlichen Krebserkrankungen das p53-Gen durch somatische Mutation geschädigt
oder ausgefallen. Als Folge häufen sich Mutationen einer Zelle, und die
Wahrscheinlichkeit für die Umwandlung normaler Zellen in Krebszellen steigt um
ein Vielfaches.
5. Reversion und Suppression von Mutationen
Bei diesen Mechanismen handelt es sich
nicht um Reparatursysteme, sondern um Rückgängigmachung oder Unterdrückung der
Mutationen. Von echter Reversion
spricht man, wenn eine Mutation an derselben Stelle durch eine Rückmutation
wieder aufgehoben wird. Am häufigsten betrifft dies Punktmutationen, Deletionen
fast nie. Diese können durch die Addition eines benachbarten Basenpaares wieder
ausgeglichen werden, da dadurch das Leseraster wieder in den korrekten
Dreiertakt kommt. Da hier nicht die genotypischen Effekte der Erstmutation
beseitigt, sondern lediglich unterdrückt werden, spricht man von Suppression. Intragenische Suppression
liegt vor, wenn die Zweitmutation auf demselben Gen liegt wie die Erstmutation,
intergenische, wenn sie auf einem
anderen Gen liegt. So kann z.B., wenn durch Mutation die Interaktion zwischen
zwei Proteinen zerstört ist, diese durch Mutation in dem zweiten Protein wieder
hergestellt werden oder es wird durch die Mutation ein zweiter Syntheseweg
aktiviert, der das zuerst mutierte Genprodukt überflüssig macht.
Weitere
Links:
Mutationen und Mutagene:
http://www.zum.de/Faecher/Materialien/beck/13/bs13-5b.htm
DNA-Reparatur:
http://www.merian.fr.bw.schule.de/Beck/skripten/11n/dnarep1.htm